Diagram:
Diagram available in:
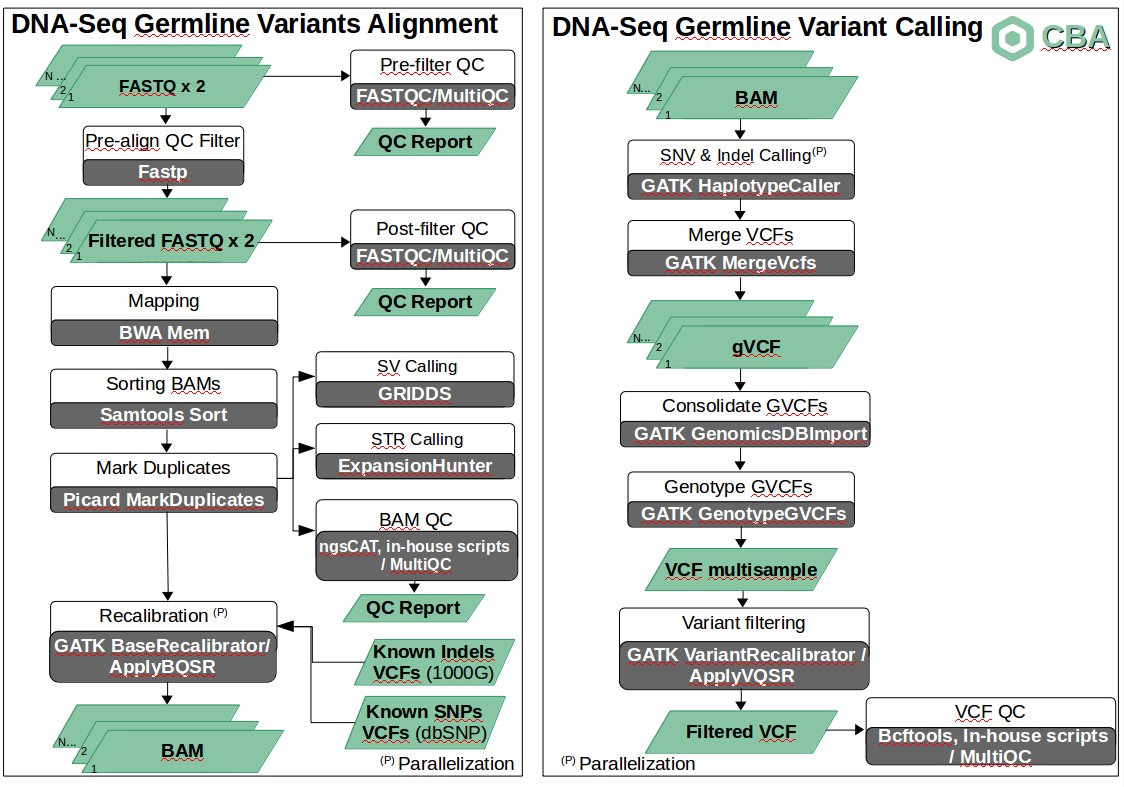
Description
The pipeline for the discovery of SNVs and small indels for WGS data is organized as a set of steps based on GATK Best Practices.
For a set of samples (cohort), the first step runs a QC study on raw FASTQ data (usually a paired-end experiment) providing a single report using FASTQC [1] and MultiQC [2] tools. If a sample has a very low quality, it is removed from the cohort. Otherwise, a QC filtering is applied so that low quality reads are removed from the original sample through fastp tool [3], obtaining a set of filtered FASTQ files. These filtered FASTQ files are then analyzed again in terms of quality (FASTQC tool), obtaining a single report for all samples through multiqc tool. For each sample and each step (pre-filter QC and pre-align QC filter) a job is run in the HPC infraestructure.
Then, each sample is aligned to the reference genome (hs37d5) by means of commonly used BWA aligner [4]. The obtained BAMs are then sorted (samtools [5]) and duplicate reads are marked (picard tools [6]). All obtained BAMs are later analyzed in terms of QC using in-house scripts, ngsCAT [7] (for targeted sequencing) and MultiQC tools. Again, for each sample, each step (mapping, sorting BAMs, marking duplicates and BAM QC) is launched as a job. At the same time and for each BAM, Structural Variants (duplications, deletions, insertions, translocations and inversions) and Short Tandem Repeats (STRs) are analyzed through GRIDDS [8] (see details here) and Expansion Hunter [9] respectively. GRIDDS is launched as a set of jobs and ExpansionHunter as a single job.
For each BAM, Base Quality Score Recalibration (BQSR) is applied in a two step procedure through GATK [10] BaseRecalibrator and ApplyBQSR tools using a set of Known Indels and SNPs, obtaining a set of BAM files ready for the identification of variants. This step is parallelized so that is run per sample and chromosome (i.e. a job per sample and chromosome).
After BQSR, variants (SNVs and small indels) are identified through GATK's HaplotypeCaller tool. For a given sample, the identification of variants is performed separately per chromosome. Once this step has finished, all gVCF files corresponding to all chromosomes for a given sample are merged in a single gVCF file by means of GATK's MergeVCFs tool, obtaining one gVCF file per sample.
After that, a joint genotyping taking into account all samples in the cohort is run though GATK GenomicsDBImport and GenotypeGVCFs tools. This way, the different records are merged together in a sophisticated manner, obtaining a single multisample VCF file.
With the aim of reducing putative false positives, a variant filtering step is then run. This step consists of a two-stage process called VQSR: the first step assigns a well-calibrated probability to each variant call in a call set and the second step consists of filtering variants based on score cutoffs identified in the first pass. For this purpose, GATK VariantRecalibrator and ApplyVQSR tools are run.
Once a filtered VCF is obtained, several statistics are generated related to the identified variants using bcftools, in-house scripts and the MultiQC tool.
[1] FASTQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
[2] Philip Ewels, Måns Magnusson, Sverker Lundin, Max Käller, MultiQC: summarize analysis results for multiple tools and samples in a single report, Bioinformatics, Volume 32, Issue 19, 1 October 2016, Pages 3047–3048
[3] Shifu Chen, Yanqing Zhou, Yaru Chen, Jia Gu; fastp: an ultra-fast all-in-one FASTQ preprocessor, Bioinformatics, Volume 34, Issue 17, 1 September 2018, Pages i884–i890
[4] Heng Li, Richard Durbin; Fast and accurate short read alignment with Burrows–Wheeler transform, Bioinformatics, Volume 25, Issue 14, 15 July 2009, Pages 1754–1760
[5] Samtools, http://www.htslib.org/
[6] Picard tools, https://broadinstitute.github.io/picard/
[7] Francisco J. López-Domingo, Javier P. Florido, Antonio Rueda, Joaquín Dopazo, Javier Santoyo-Lopez; ngsCAT: a tool to assess the efficiency of targeted enrichment sequencing, Bioinformatics, Volume 30, Issue 12, 15 June 2014, Pages 1767–1768
[8] Daniel L. Cameron, Jan Schröder, Jocelyn Sietsma Penington, Hongdo Do, Ramyar Molania, Alexander Dobrovic, Terence P.Speed, Anthony T.Papenfuss; GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly, Genome Research, 27(12), 2017, pp-2250-2060
[9] Egor Dolzhenko, Viraj Deshpande, Felix Schlesinger, Peter Krusche, Roman Petrovski, Sai Chen, Dorothea Emig-Agius, Andrew Gross, Giuseppe Narzisi, Brett Bowman, Konrad Scheffler, Joke J F A van Vugt, Courtney French, Alba Sanchis-Juan, Kristina Ibáñez, Arianna Tucci, Bryan R Lajoie, Jan H Veldink, F Lucy Raymond, Ryan J Taft, David R Bentley, Michael A Eberle; ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions, Bioinformatics, vol.35, issue 22, 2019, pages 4754-4756
[10] Ryan Poplin, Valentin Ruano-Rubio, Mark A. DePristo, Tim J. Fennell, Mauricio O. Carneiro, Geraldine A. Van der Auwera, David E. Kling, Laura D. Gauthier, Ami Levy-Moonshine, David Roazen, Khalid Shakir, Joel Thibault, Sheila Chandran, Chris Whelan, Monkol Lek, Stacey Gabriel, Mark J. Daly, Benjamin Neale, Daniel G. MacArthur, Eric Banks; Scaling accurate genetic variant discovery to tens of thousands of samples, bioRxiv 201178; doi: https://doi.org/10.1101/201178