This page describes the process followed for the determination and analysis of SVs for germ or somatic samples. It is specially adapted for illumina paired-end sequencing, both of exomes and of complete genomes.
This pipeline is based on GRIDSS1 best practices:
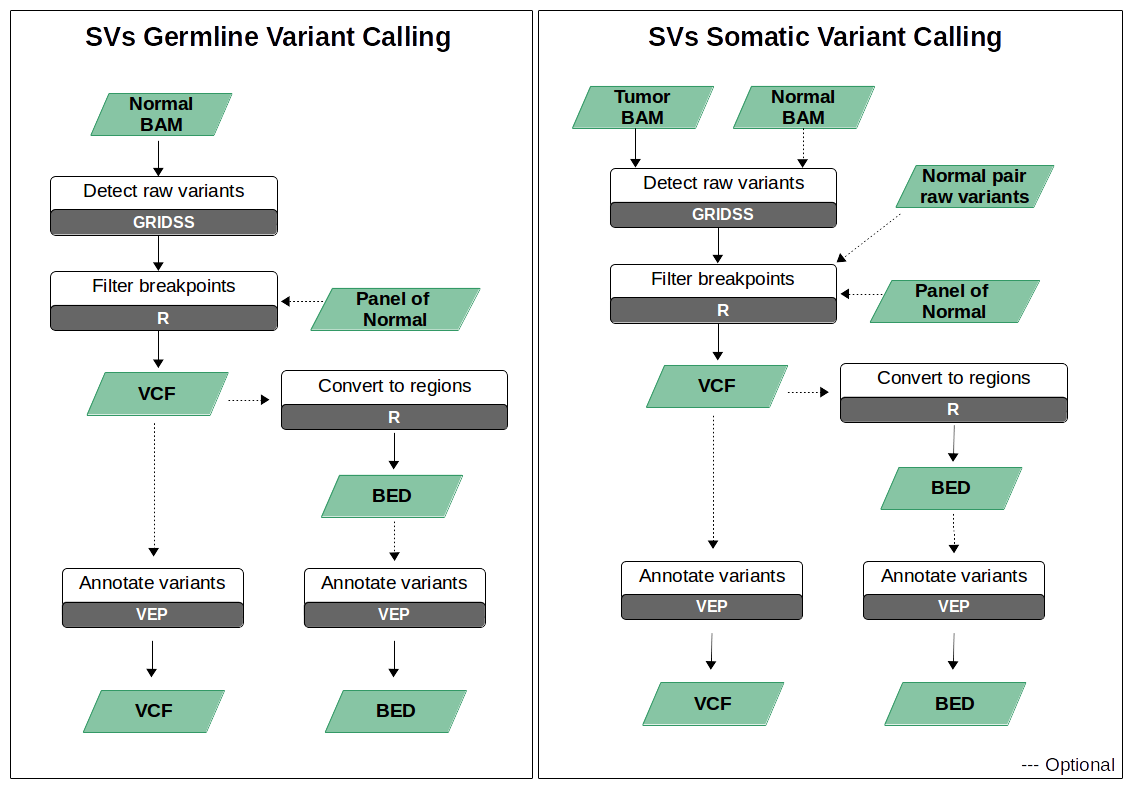
General Pipeline Structure
The pipeline has been developed using the WDL workflow development language. The implemented WDL code is specifically prepared to be executed by using the Cromwell tool. To facilitate execution, the pipeline has been encapsulated within a bash script that automates each of the steps. The complete pipeline is represented in the following diagram:
Diagram available in: sv_workflow_diagram.odp
Requirements and recommendations
This pipeline is specifically developed for illumina paired-end sequencing, although it could work with other technologies.
To filter raw variants, it is recommendable to create a panel of normal (PoN). It is recommended that the PoN be formed with at least 30 independent samples sequenced under the same conditions as the test sample.
It is recommended to use BAMs with duplicate reads marked or, in the case of PCR-free, unmarked.
It is recommended not to eliminate BAM reads (duplicates, low quality by alignment, etc.)
It is not advisable to call on regions with low mapping, segmental duplication or high GC content.
[1] Cameron DL, Schröder J, Penington JS, Do H, Molania R, Dobrovic A, Speed TP, Papenfuss AT. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Research, 2017 Dec;27(12):2050-2060. https://github.com/PapenfussLab/GRIDSS