Mechanistic models for drug repurposing in rare diseases with machine learning methods
Funding

With the increasingly vast trend of data generation, machine learning (ML) methodologies are consolidating as ideal tools to extract new information from Big Data repositories beyond the original purpose that generated these repositories. Of special importance are fast growing public genomic Big Data repositories, given that they potentially contain keys for the genetic basis of numerous traits of medical relevance.
The possibility of using genomic repositories to formulate hypotheses and extrapolate conclusions to fields in which little biological knowledge is available, i.e. Rare Diseases (RD), constitutes an attractive opportunity for genomic Big Data analysis. RDs constitute an important health challenge because of the RD paradox: despite individually any RD has a low prevalence, the existence of more than 6000 RDs results in a total incidence of 6% in the population (similar to those of many common diseases). Unfortunately only 400 of them have an effective treatment, essentially because they are not contemplated by pharma companies as interesting market niches.
Therefore, an innovative strategy to apply to RD drug discovery is the repurposing of drugs with indications already approved for other diseases. In this project we will systematically develop mathematical mechanistic models of for about 100 RDs that will be used to predict the effect of therapeutic interventions, select potential therapeutic targets that revert disease to the normal status or alleviate its symptoms and, among the targets found, select those that are already targets for known drugs. Information from signaling and metabolic pathways will be used as a starting point to define functional outcomes of diseases and to connect other disease genes to them.
ML methods will be used to extract potential relationships between disease genes in order to build comprehensive disease maps. However, in spite of the abundance of genomic data, the number of variables to take into account is also high, which makes ML methods prone to the course of dimensionality, leading to unstable solutions from an interpretability point of view. To overcome these problems we will make an innovative use of regularization and variational approximations coupled with feature ranking stability methods in genomics, which are less prone to overfit the data, leading to trustworthy models.
ML analysis of genomic Big Data will definitely change the paradigm of research in RD, where diseases are typically addressed one at a time. ML analysis of genomic Big Data will definitely change the paradigm of research in RD, where diseases are typically addressed one at a time. This proposal constitutes an innovative approach to domains in which no much information exists but genomic Big Data are available, by using ML to expand the current biological knowledge.
Ellis-Van Creveld syndrome (EVC) is a congenital skeletal and ectodermal dysplasia, characterized by short limb dwarfism, additional fingers and/or toes (polydactyly), abnormal development of fingernails and congenital heart defects.
Ellis–van Creveld syndrome often is the result of founder effects in isolated human populations, such as the Amish and some small island inhabitants. Observation of the inheritance pattern has illustrated that the disease is autosomal recessive. Ellis–van Creveld syndrome is caused by a mutation in the EVC gene, located in chromosome 4 short arm and which function is not well understood at this time, as well as in EVC2, located close to the EVC gene.
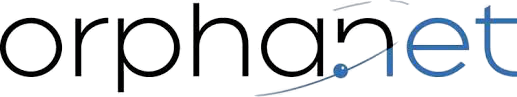
Familial melanoma (FM) is a rare inherited form of melanoma, a type of skin cancer that develops from the pigment-producing cells known as melanocytes. FM is characterized by development of melanoma in two first degree relatives or more relatives in an affected family and accounts for about 10% of all cases of cutaneous melanoma.
The risk of familial melanoma is closely related to a wide range of genetic alterations in susceptibility genes but also appears to be influenced by phenotypic risk factors. The most common high-penetrance susceptibility gene implicated in FM is CDKN2A, accounting for predisposition in approximately 20% of FM. CDK4, another high risk gene, is rarely involved. Mutations of BAP1,POT1, TERF2IP, ACD,and TERT have recently been reported and penetrance remains to be determined. Medium penetrance genes include MC1R.

Fanconi anaemia (FA) is a rare genetic disease resulting in impaired response to DNA damage. Among those affected, the majority develop cancer, most often acute myelogenous leukemia, and 90% develop bone marrow failure (the inability to produce blood cells) by age 40. About 60–75% of people have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% of people have some form of endocrine problems, with varying degrees of severity.
FA is due to mutations in genes involved in DNA repair and genomic stability and it presents a recessive inheritance pattern. Fifteen genes representing 15 complementation groups have been identified.
Hirschsprung's disease (HD or HSCR) is a birth defect characterized by signs of intestinal obstruction due to nerves missing from parts of the intestine. Symptoms include constipation, vomiting, abdominal pain, diarrhea and failure to thrive. Complications may include enterocolitis, megacolon, bowel obstruction and intestinal perforation. Typically, Hirschsprung disease is diagnosed shortly after birth, although it may develop well into childhood.
Genetic and environmental factors play a role in its pathogenesis. Several genes are associated with HSCR, particularly: the Ret proto-oncogene (RET), the glial cell derived neurotrophic factor gene (GDNF), the neurturin gene (NRTN), the endothelin B receptor gene (EDNRB), the endothelin-3 gene (EDN3), the endothelin-converting enzyme 1 gene ECE1, and the L1 cell adhesion molecule gene L1CAM.
Albinism is a group of congenital disorders characterized in humans by the complete or partial absence of pigment in the skin, hair and eyes. Oculocutaneous albinism (OCA) is associated with a number of vision defects, such as photophobia, nystagmus, and amblyopia. Lack of skin pigmentation makes for more susceptibility to sunburn and skin cancers.
Albinism results from inheritance of recessive gene alleles and is known to affect all vertebrates. In humans, it is due to absence or defect of tyrosinase, a copper-containing enzyme involved in the production of melanin. Variants include OCA1A (the most severe form), OCA1B, OCA1-minimal pigment (OCA1-MP), OCA1-temperature sensitive (OCA1-TS), OCA2, OCA3, OCA4, OCA5, OCA6 and OCA7.

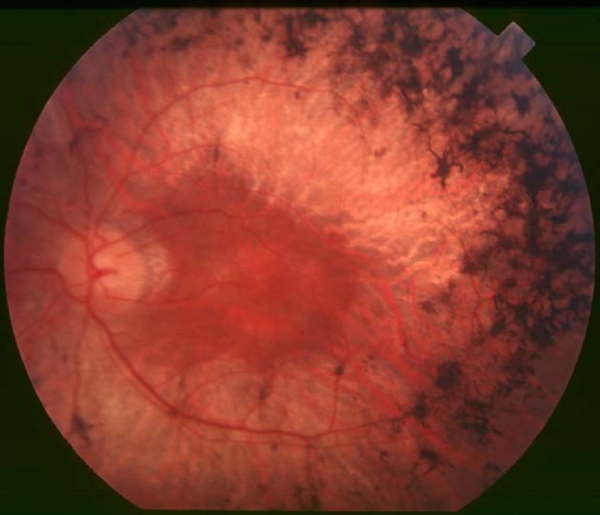
Retinitis pigmentosa (RP) is an inherited retinal dystrophy leading to progressive loss of the photoreceptors and retinal pigment epithelium and resulting in blindness usually after several decades. Retinitis pigmentosa is slowly progressive but relentless. There is however broad variability in age of onset, rate of progression and secondary clinical manifestations. Symptoms include trouble seeing at night (nyctalopia) and decreased peripheral vision (side vision). As peripheral vision worsens, people may experience "tunnel vision" that can end in blindness. Mutations in one of more than 50 genes are involved.
Williams syndrome is caused by a deletion of about 27 genes from the long arm of chromosome 7s, manner. It is an autosomal dominant genetic disorder that affects many parts of the body. Facial features are characteristic, presenting an appearance that has been described as "elfin". While mild to moderate intellectual disability, in particular problems with visual spatial tasks such as drawing, verbal skills are generally relatively unaffected. Those affected often have an outgoing personality, are extremely social and appear happy. Problems with teeth, heart problems (especially supravalvular aortic stenosis), and periods of high blood calcium are the most common symptoms.
Esteban-Medina, M., Peña-Chilet, M., Loucera, C. et al. Exploring the druggable space around the Fanconi anemia pathway using machine learning and mechanistic models. BMC Bioinformatics 20, 370 (2019).
https://doi.org/10.1186/s12859-019-2969-0
Montanuy H, Martínez-Barriocanal Á, Antonio Casado J, et al. Gefitinib and Afatinib Show Potential Efficacy for Fanconi Anemia-Related Head and Neck Cancer [published online ahead of print, 2020 Jan 31]. Clin Cancer Res. 2020;10.1158/1078-0432.CCR-19-1625.
https://doi.org/10.1158/1078-0432.CCR-19-1625